ARTICULO PARA COLEGAS
MiopatÃas Inflamatorias Idiopáticas: un desafÃo diagnóstico
APORTES DEL LABORATORIO
Las miopatÃas inflamatorias idiopáticas (MII) son un conjunto de enfermedades heterogéneas, crónicas, de etiologÃa desconocida, cuya principal caracterÃstica es la debilidad muscular y la identificación de una inflamación en la biopsia muscular. La presencia de autoanticuerpos especÃficos y asociados a estas enfermedades sustenta la etiologÃa autoinmune del proceso y ayuda a categorizar a los pacientes.Â
Dentro de este grupo las entidades más representativas son polimiositis (PM) y dermatomiositis (DM) del adulto, formas juveniles de miopatÃa inflamatoria principalmente DM, miositis por cuerpos de inclusión (MCI), miopatÃa asociada a cáncer (MCA). También se incluyen miopatÃa necrotizante inmunomediada (MNIM), miositis de los sÃndromes de superposición y sÃndrome antisintetasa (SAS). Se ha reconocido un subgrupo de DM en el que las manifestaciones cutáneas se presentan durante perÃodos prolongados en ausencia de enfermedad muscular, denominándose DM sine miositis o DM amiopática.
Se consideran enfermedades sistémicas, especialmente DM y PM, ya que aunque el principal órgano diana es el músculo estriado, otras estructuras, como la piel o el sistema articular, se afectan con frecuencia. También los órganos internos, en especial el pulmón, forman parte del espectro clÃnico de estas enfermedades. Ocasionalmente, y sobre todo la dermatomiositis, puede asociarse a cáncer, y presenta un comportamiento paraneoplásico.
Incidencia: Las MII presentan una incidencia anual de 0,8-8 casos/millón de habitantes/año y una prevalencia de 5-8 casos/100.000 habitantes. Es más frecuente en mujeres (2:1).
Pueden presentarse a cualquier edad, con dos picos de mayor incidencia: uno en la infancia (10-15 años) que corresponde a la DM juvenil, y otro en la edad adulta (45-60 años). La miositis por cuerpo de inclusión (MCI) es más común después de los 50 años y predominantemente en varones.
Etiologia : es desconocida, aunque se ha sugerido la implicancia de un agente externo fÃsico, quÃmico o infeccioso que actúa en un territorio genéticamente predispuesto.
Se observa una alteración de la inmunidad tanto celular como humoral. Prueba de ello es el acúmulo de linfocitos en el tejido muscular, la presencia de autoanticuerpos especÃficos dirigidos contra moléculas citoplasmáticas implicadas en la sÃntesis de proteÃnas y la respuesta a agentes inmunosupresores.
Es probable que la respuesta de autoanticuerpos en las MII esté controlada por antÃgeno y genéticamente determinada por alelos HLA de clase II. Los principales marcadores alélicos asociados con la MII son B*0801 y DRB1*0301.
El tratamiento incluye la administración de glucocorticoides, inmunodepresores y terapias biológicas.
MANIFESTACIONES CLÃNICAS
La forma de presentación más común de estas enfermedades es la debilidad muscular, que suele afectar de forma caracterÃstica a la musculatura esquelética proximal, es decir a la cintura escapular y pelviana, dificultando las actividades que precisan del normal funcionamiento de estos músculos, como tender la ropa, peinarse, subir escaleras o levantarse de la silla, entre otras. Los músculos flexores del cuello y la musculatura estriada de la orofaringe se afectan con frecuencia, estos últimos causando dificultad en la deglución y en la respiración.
El compromiso cutáneo precede en unos 6 meses a la enfermedad muscular. Las manifestaciones cutáneas son caracterÃsticas de la dermatomiositis, y podemos distinguir un amplio abanico de lesiones, la mayorÃa de ellas con un cierto componente de fotosensibilidad, por lo que suelen aparecer en zonas expuestas al sol. Se consideran patognomónicas las pápulas de Gottron, áreas eritematosas discretamente descamativas e infiltradas que aparecen sobre los nudillos de las manos (80 % de los pacientes) y el edema palpebral de color lila o en heliotropo (60 % de los pacientes). Lesiones similares pueden observarse en zonas de extensión, como codos y rodillas, y también en la lÃnea de inserción del cuero cabelludo y en la nuca.
Otros hallazgos cutáneos incluyen el eritema en cara, cuello y pecho (signo de V), o en la parte posterior del cuello y hombros (signo del chal), la hiperqueratosis de los dedos (dedos de carpintero o de mecánico) y las telangiectasias periungueales.
Las manifestaciones pulmonares son la manifestación visceral más frecuente, aparece en hasta casi la mitad de los pacientes en una u otra forma. Se manifiesta en forma de enfermedad pulmonar intersticial (EPI), hipoventilación de origen miopático o neumonÃa por aspiración. Los pacientes con DM-PM y EPI tienen una mayor mortalidad y peor pronóstico que aquellos que no la presentan. Los sÃntomas más frecuentes que hacen pensar en una EPI son la tos y la disnea, si bien hasta 1/3 de los pacientes pueden ser asintomáticos.
A nivel gastrointestinal la miositis farÃngea y del tracto superior del esófago puede ocasionar disfagia proximal tanto a sólidos como lÃquidos y regurgitación nasal de lÃquidos. También puede provocar voz nasal y ronquera asà como neumonÃas por aspiración.
La afectación cardÃaca es frecuente, pero suele cursar de forma asintomática hasta fases evolucionadas de la enfermedad. Las alteraciones más frecuentes son los defectos de conducción y las arritmias, aunque se han descrito casos de miocarditis, fibrosis miocárdica, miocardiopatÃa dilatada, vasculitis coronaria (ángor e infarto de miocardio), pericarditis y taponamiento pericárdico.
DIAGNÃSTICO
Realizar el diagnóstico de las MII suele ser un desafÃo. Conjuntamente con la anamnesis y examen fÃsico que incluye la evaluación de fuerza muscular y la búsqueda de manifestaciones extra musculares, son de utilidad las pruebas de laboratorio como la determinación de las enzimas musculares y el laboratorio inmunológico, los estudios de imágenes no invasivos como RM de músculo y TomografÃa Computada de tórax y estudios invasivos como el electromiograma (EMG) y la biopsia muscular.
LABORATORIO
Enzimas: El daño muscular ocasiona un aumento de los niveles de enzimas musculares. La elevación de la creatin fosfokinasa (CPK) es el indicador más sensible y especÃfico de la enfermedad muscular activa, presente en el 90% de pacientes. La aldolasa sérica es un indicador menos sensible que la CPK para detectar la miositis activa, sin embargo, sus niveles pueden estar elevados en presencia de CPK normal. Las transaminasas hepáticas (GOT, GPT) y la lactato deshidrogenasa (LDH) pueden estar elevadas, pero son poco especÃficas.
La determinación de las enzimas musculares es importante para el diagnóstico de la enfermedad, para el monitoreo de la respuesta al tratamiento y/o para detectar posibles recidivas.
Autoanticuerpos:Â Como en otras enfermedades del tejido conectivo (ETC), se detectan, por inmnunofluorescencia indirecta (Hep2), anticuerpos antinucleares (FAN/ANA) en un 50 a 80 % de los pacientes con miopatÃa inflamatoria, especialmente en PM y DM.
En la MCI la positividad es solo del 20%. En las miositis asociadas a otras ETC la frecuencia es más alta y la más baja se presenta en las miositis asociadas a malignidad. Los ANA solo tienen valor como guÃa diagnóstica, permitiendo en tÃtulos altos diferenciar las MII de las distrofias o miopatÃas no autoinmunes.
Frente a un resultado positivo de FAN y sospecha clÃnica de MII se debe proseguir el estudio con técnicas de inmunodetección especÃfica para antÃgenos.
En los últimos años dentro del campo de las MII se han reconocido y caracterizado un gran número de autoanticuerpos, se clasifican en dos grandes grupos: anticuerpos especÃficos de miositis (AEM) y anticuerpos asociados a miositis (AAM).
Los anticuerpos especÃficos de miositis (AEM): solo se presentan en este grupo de enfermedades. Son considerados altamente especÃficos para MII y la estrecha correlación de éstos con el fenotipo clÃnico y su papel en el pronóstico, ha llevado al desarrollo de sistemas de clasificación clÃnico-serológicos.
Dentro de este grupo tenemos los anticuerpos antisintetasas y los no antisintetasa
ANTICUERPOS ESPECÃFICOS DE MIOSITIS
| Autoanticuerpos especÃficos | AntÃgeno | Frecuencia (%) | Fenotipo clÃnico |
|---|---|---|---|
| Anticuerpos antisintetasas | |||
| Anti-Jo-1 Anti-PL-7 Anti-PL-12 Anti-OJ Anti-EJ Anti-KS Anti-Z0 Anti-HA |
Histidil-tRNA-sintetasa Treonil-tRNA-sintetasa Alanil-tRNA-sintetasa Isoleucil-tRNA-sintetasa Glicil-tRNA-sintetasa Asparagil-tRNA-sintetasa Fenilalanil-tRNA-sintetasa Tirosil-tRNA-sintetasa  Son enzimas citoplasmáticas que catalizan la unión covalente de los aminoácidos con su tRNA |
10-40 5-10 <5 <5 5-10 <1 <1 <1 |
SÃndrome antisintetasa: miositis, artritis, afección pulmonar, fiebre, manos de mecánico y fenómeno de Raynaud. |
| Anticuerpos no antisintetasa | |||
| Anti-SRP | PartÃcula de reconocimiento de la señal. Complejo citoplasmático que media la traslocación de polipéptidos a través del retÃculo endoplasmático. |
4-5 | PM de comienzo muy agudo y grave, afección muscular (miopatÃa necrosante), cardÃaca y disfagia, escasa afección pulmonar. |
| Anti-Mi-2 | Helicasa nuclear | 5-14 | Comienzo agudo y leve, lesiones cutáneas caracterÃsticas de DM, poca afección pulmonar. Sensibilidad a los esteroides. |
| Anti-p155/140 (TIF-1 gamma) |
Factor intermediario transcripcional 1 gamma |  | DM asociada a cáncer Formas juveniles de DM |
| Anti-NXP2 | ProteÃna nuclear de la matriz |  | Mayor incidencia de calcinosis, menor edad de inicio, buena respuesta a la terapia. Más frecuente en DM juvenil. DM asociada a cáncer. |
| Anti-MDA5 (Anti-CADM-140) |
ProteÃna 140 kDa | 50 en DMA | DM amiopática: compromiso muscular ausente o mÃnimo, alta incidencia de EPI. |
| Anti-HMGCR | HMGCR (HMG CoA reductasa) | Â | MiopatÃa necrotizante relacionada en la mayorÃa de los casos a la ingesta de estatinas. Resistente a la terapia. |
| Anti-SAE1 | SAE (enzima activadora-modificadora de small ubiquitin-like) |  | DM: miositis asociada a cáncer, DM con EPI rápidamente progresiva. |
Sólo en un tercio de los pacientes se detectan anticuerpos especÃficos de miositis.
Estos otorgan información para el diagnóstico, permiten clasificar a los pacientes en diferentes subtipos clÃnicos, evaluar su pronóstico, riesgo de cáncer y posible respuesta al tratamiento.
De los AEM los más relevantes son:
- los anticuerpos antisintetasa. Los pacientes con anticuerpos antisintetasa tienen una mayor frecuencia de enfermedad pulmonar intersticial (95%) respecto a los pacientes negativos (40%), predicen una mejor respuesta inicial al tratamiento con esteroides, aunque se asocian con una mayor tasa de recidivas. Pueden incluso anteceder a la aparición de miopatÃa.
Definen un grupo de pacientes con caracterÃsticas clÃnicas comunes: sÃndrome antisintetasa caracterizado por miositis, enfermedad pulmonar intersticial difusa, Fenómeno de Raynaud, manos de mecánico y artritis.
De estos anticuerpos, el anti Jo-1 es el más frecuente. Su presencia se asocia a un curso clÃnico severo y peor pronóstico. El tÃtulo fluctúa con la actividad de la enfermedad y puede desaparecer con el tratamiento.
- De los anticuerpos no antisintetasa, se destaca el anti-SRP que provoca un cuadro severo de polimiositis, de carácter agudo con miopatÃa necrotizante y resistente al tratamiento.
El anti-Mi-2, aparece fundamentalmente en pacientes con dermatomiositis, incluyendo formas juveniles.
- Los anticuerpos asociados a miositis (AAM), no son especÃficos de las miopatÃas, pueden aparecer en otras colagenopatÃas y sÃndromes de superposición.
ANTICUERPOS ASOCIADOS A MIOSITIS
| Anticuerpo especifico | AntÃgeno | Frecuencia (%) | Fenotipo ClÃnico |
| Anti-U1-RNP | U1RNP nuclear (proteÃnas A 34 KDa, C 33 KDa, 70 KDa) | 10 | Enfermedad del tejido conectivo (EMTC) |
| Anti-PM/Scl | Complejo nucleolar (14-16 proteÃnas) con función exorribonucleasa durante el procesamiento del ARN | 5-25 | SÃndrome de solapamiento: miostis/esclerodermia |
| Anti SSA (Ro 52 y Ro 60) | ProteÃnas del RNA | 10-25 | Miositis con Lupus Eritematoso Sistémico o SÃndrome de Sjogren |
| Anti-Ku | ProteÃnas del ADN | 10-25 | SÃndrome de solapamiento: miositis-LES/esclerodermia |
| snRNP no U1 | U2, U4/6, U5 RNP | Â | SÃndrome de solapamiento: miositis-esclerodermia |
Aunque estos autoanticuerpos no son especÃficos para miositis, su presencia puede ser útil para distinguir una miopatÃa inflamatoria de una miopatÃa no autoinmune.
Los anticuerpos anti-PM/Scl y anti-SSA/Ro son los más frecuentes de este grupo. El anti- PM/Scl es detectado en un 24 % de pacientes con sÃndrome de solapamiento miositis/esclerodermia. Es un marcador de buen pronóstico. Los autoantÃgenos dentro del complejo han sido identificados como PM-Scl75 y PM-Scl100. La presencia en PM o DM del anticuerpo anti-PM-Scl se asocia a afectación pulmonar y esofágica.
El anti SSA/Ro 52 KDA es detectado en MII asociado a anticuerpos antisintetasa en un 70 % de los pacientes.
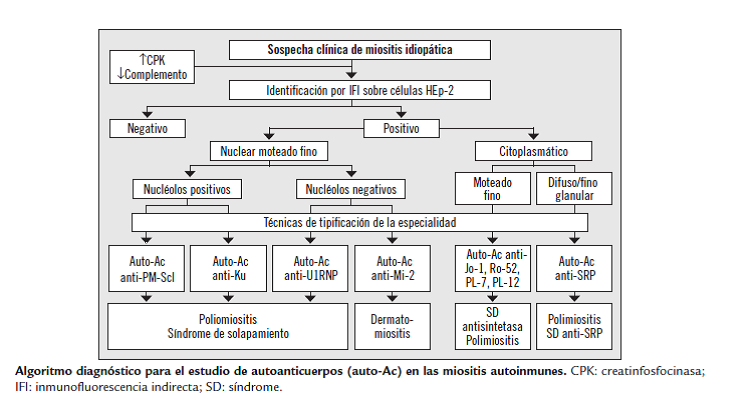
En la evaluación inicial entonces de todo paciente con sospecha de MII se debe incluir la determinación de las enzimas musculares, medición de los niveles de complemento, reactantes de fase aguda y el estudio de autoanticuerpos : FAN, anti-ADN, anti-Ro, anti-La, anti-Sm, anti-RNP.
El laboratorio cuenta con la posibilidad de realizar un Panel Ampliado de Anticuerpos IGG para completar el abordaje de las miopatias.
MIOSITIS - PANEL AMPLIADO AC IGG
Método : LIA - Inmunoensayo Lineal
Mi-2 alfa Ac ................:
Mi-2 beta Ac ................:
TIF1 gamma Ac ...............:
MDA5 Ac .....................:
NXP2 Ac .....................:
SAE1 Ac .....................:
Ku Ac .......................:
PM-Scl 100 Ac ...............:
PM-Scl 75 Ac ................:
Jo-1 Ac .....................:
SRP Ac ......................:
PL-7 Ac .....................:
PL-12 Ac ....................:
EJ Ac .......................:
OJ Ac .......................:
Ro-52 Ac ....................:
Tipo de muestra: Suero
Dias de proceso: Lunes a viernes
Resultados: 20 dias habiles
El incremento de los niveles de las enzimas musculares y la presencia de autoanticuerpos son los datos de laboratorio más caracterÃsticos de las MII, constituyendo una herramienta que brinda información clÃnicamente útil para el diagnóstico y seguimiento de los pacientes con MII. El diagnóstico temprano es crucial para el inicio oportuno del tratamiento.
BibliografÃa
-
MiopatÃas inflamatorias. Dermatomiositis, polimiositis y miositis con cuerpos de inclusión. Albert Selva O?Callaghan y Ernesto Trallero Araguás. Reumatol Clin. 2008; 4(5):197-206.
-
Métodos de diagnóstico en el estudio de las MiopatÃas Inflamatorias Autoinmunes. Revista Argentina de ReumatologÃa 2020; 31(1):3 -7.
-
MiopatÃas autoinmunes: revisión de diagnóstico y manejo. An Fac med. 2019; 80(3):362-71.
-
El laboratorio en el estudio de las miopatÃas inflamatorias idiopáticas. Gargiulo MarÃa de los Angeles, Pérez Nicolás, Khoury Marina, Suarez Lorena, Collado MarÃa Victoria, Ãlvarez Mónica, Gómez Graciela. B y PC 2020; 84(2):20-25.
-
MiopatÃas inflamatorias idiopáticas: una mirada actualizada al diagnóstico y el manejo. Rev. méd. Chile vol.147 no.3 Santiago mar. 2019.
-
Autoantibodies in myositis. Nature reviews Reumatology. Vol. 14 mayo 2018.
Dra. Ãrica RodrÃguez
Departamento de InmunologÃa
Dra. Delia Ostera
Departamento de Neurociencias