ARTICULO PARA COLEGAS
Leucemia Mieloide Crónica
La leucemia mieloide crónica (LMC) es un sÃndrome mieloproliferatvo crónico de naturaleza clonal, con origen en una célula madre pluripotencial común a las 3 series hematopoyéticas, en las cuales se encuentra presente el cromosoma Filadelfia (Ph). Este es el cromosoma 22 acortado producto de una translocación recÃproca t(9;22)(q34,q11), en donde se adiciona el segmento 3´ del gen ABL al segmento 5´ del gen BRC, creando un gen hÃbrido BCR-ABL que da lugar a un transcripo de ARNm quimérico de 8,5kb.
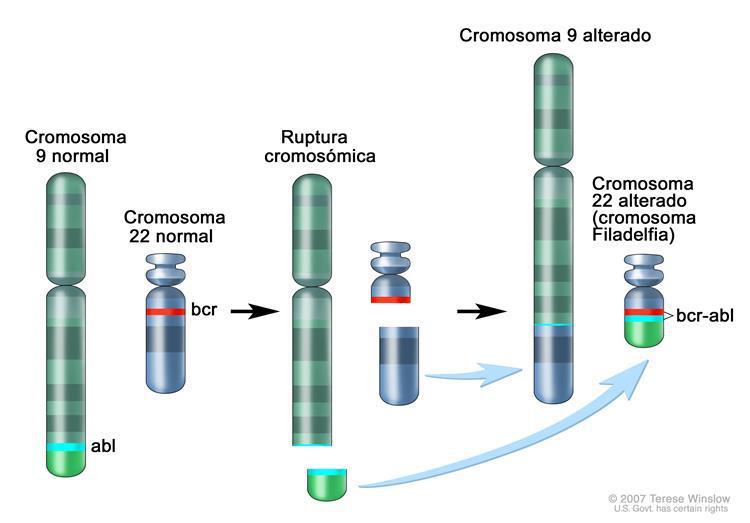
Esta enfermedad, que representa del 15 al 20 % del total de leucemias tiene una edad media de aparición de alrededor de 53 años y la incidencia máxima es entre los 30 y los 40. La translocación BCR-ABL se puede encontrar en el 95% de todos los casos de LMC.
Alrededor de la mitad de los pacientes son asintomáticos y su diagnóstico es un hallazgo al observar un recuento de leucocitos elevado en un hemograma (hiperleucocitosis). Sin embargo hay un 50% de pacientes que sà presentan sÃntomas como astenia, anorexia, pérdida de peso y esplenomegalia.
Esta leucemia se divide en tres fases, la primera que es la fase crónica, es un estado indolente en el que se diagnostica al 90% de los pacientes y se caracteriza por una expansión de células mieloides con una maduración normal. La segunda fase que se denomina acelerada es cuando comenzamos a detectar en sangre periférica células sin diferenciar. Y por último la enfermedad termina con la crisis blástica, en la cual evidenciamos más de un 30% de blastos en sangre periférica e infiltrados extramedulares.
El gen hÃbrido se puede formar de distintas maneras. Mientras que el punto de ruptura del gen ABL puede ocurrir en cualquier sitio a lo largo de una zona de 300kb en un extremo 5, el gen BCR tiene tres regiones diferentes de ruptura. El primer sitio de ruptura se encuentra en un área de 5,8kb entre los exones 12 y 16, llamada región de ruptura mayor (M-BCR), la cual codifica dos transcriptos de fusión e13a2 (b2a2) y e14a2 (b3a2), que dan orÃgen a una proteÃna quimérica de 210kDa caracterÃstica de los paciente con LMC, y también puede encontrarse en una tercera parte de los pacientes que padecen leucemia linfoblástica aguda (LLA). El segundo sitio de ruptura del BCR se encuentra en el exón 2, en un área de 5,4 kb conocida como región de ruptura menor (m-BCR) y codifica una proteÃna de 190 kDa codificado por el transcripto e1a2, presente generalmente en pacientes con LLA. El último sitio de ruptura es conocido como ?-BCR y se ubica en el exón 19 del BCR (transcripto e19a2), codificando una proteÃna de 230 kDa caracterÃstica de leucemia neutrofÃlica crónica.
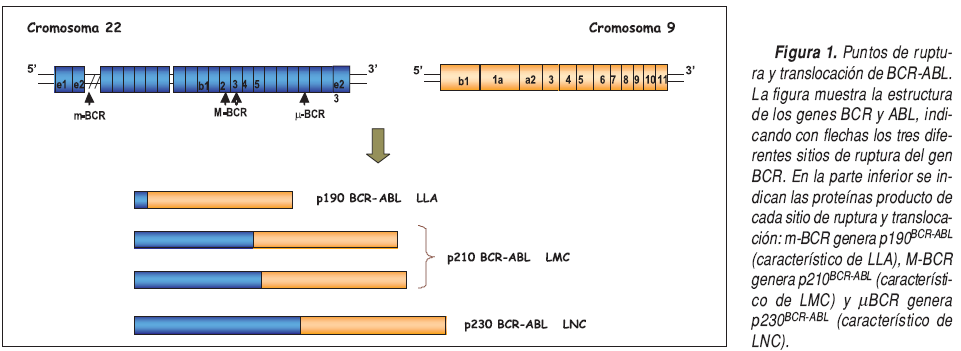
Este gen hÃbrido y por consiguiente la proteÃna quimérica BCR-ABL es la responsable del fenotipo leucémico ya que la actividad tirosin-quinasa es central para la transformación de células madre hematopoyéticas, en las cuales se produce la activación de múltiples señales de las vÃas de traducción lo que conduce a un incremento de la proliferación, reduce la dependencia de los factores de crecimiento y la apoptosis, asà como una interacción anormal entre la matriz extracelular y el estroma medular. Todos estos cambios le brindan a las células afectadas una ventaja en el crecimiento y la sobrevida sobre las células normales, lo cual conduce a las manifestaciones clÃnicas de la LMC.
El curso indolente de la LMC no elimina el requerimiento de tratamiento inmediato. El busulfán, la hidroxiurea e Interferón-?, han sido usados tradicionalmente en el tratamiento de la fase crónica, aunque en años recientes el manejo de drogas generadas por diseño (Imatinib, Dasatinib y Nilotinib) ha cambiado la historia natural de la enfermedad, asà como la calidad de vida de los pacientes. En diciembre de 2002, la FDA aprobó el Imatinib como tratamiento de primera lÃnea en pacientes con LMC. El éxito de esta droga se debe a que es un inhibidor de tirosin-quinasa que inhibe competitivamente en el sitio de unión del ATP de la proteÃna de fusión.
Para poder medir la eficiencia de los tratamientos se utilizan distintos criterios que incluyen parámetros hematológicos, citogenéticos y moleculares.
Se dice que un paciente se encuentra en remisión hematológica completa cuando presenta un recuento normal de células sanguÃneas, asà como la desaparición completa de signos y sÃntomas de la enfermedad.
A la respuesta citogenética completa se llega cuando hay ausencia total de metafases Ph+, empleando ensayos de citogenética clásica. Una respuesta citogenética parcial implica la presencia de 1 a 35% de metafases Ph+.
La remisión molecular completa indica que no existen transcriptos BCR-ABL detectables por técnicas moleculares altamente sensibles. Estas técnicas permiten el monitoreo de pacientes en tratamiento con las drogas dirigidas a inhibir la actividad de tirosin-quinasa. A través de la PCR en tiempo real podemos cuantificar la cantidad de transcriptos a lo largo del tratamiento y asà controlar que el mismo funcione. En algunos estudios se ha comprobado que luego de un determinado perÃodo en el que el paciente se encuentra en remisión molecular completa es factible suspender los inhibidores de tirosin-quinasa y aun asà un alto porcentaje no vuelve a presentar transcriptos BCR-ABL en los controles posteriores. En el caso de que vuelvan a aparecer dichos transcriptos la medicación puede ser retomada nuevamente, y se ha evidenciado que vuelve a funcionar.
Referencias
- S Branford, NCP Cross, A Hochhaus, J Radich, G Saglio, J Kaeda, J Goldman and T Hughes. Rationale for the recommendations for harmonizing current methodology for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia (2006) 20, 1925â1930.
- J Gabert, E Beillard, VHJ van der Velden, W Bi, D Grimwade, N Pallisgaard, G Barbany, G Cazzaniga, JM Cayuela, H Cave, F Pane, JLE Aerts, D De Micheli, X Thirion, V Pradel, M González, S Viehmann, M Malec, G Saglio and JJM van Dongen. Standardization and quality control studies of âreal-timeâ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia â A Europe Against Cancer Program. Leukemia (2003) 17, 2318â2357
- MarÃa Antonieta Chávez-Gonzalez, Manuel Ayala-Sánchez, Héctor Mayani. La leucemia mieloide crónica en el siglo XXI: biologÃa y tratamiento. Rev Invest Clin 2009; 61 (3): 221-232.
- Dra. Valia Pavón Morán, Dr. Porfirio Hernández RamÃrez, Dra. Gisela MartÃnez Antuña, Dra. Olga Agramonte Llanes, Dr. Juan Carlos Jaime Fagundo y Lic. Jeny Bravo Regueiro. Leucemia mieloide crónica. Actualización en Citogenética y BiologÃa Molecular. Revista Cubana de HematologÃa, InmunologÃa y Hemoterapia, versión On-line ISSN 1561-2996, ciudad de la Habana Mayo-Ago 2005.
- Jorge Cruz-Rico, Osvaldo Garrido-Acosta, Liliana Anguiano-Robledo, Ulises RodrÃguez-Wong, Elizabeth Pérez-Cruz, Jaime Sánchez-Navarrete, Nancy Jannet Ruiz-Pérez, MarÃa del RocÃo Montes-Vera. Imatinib: farmacocinética. Re Hosp Jua Mex 2013; 80(1): 67-72.
- A Hochhaus, T Masszi, FJ Giles, JP Radich, DM Ross, MT Gómez Casares, A Hellmann, J Stentoft, E Conneally, V GarcÃa-Gutiérrez, N Gattermann, W Wiktor-Jedrzejczak, PD le Coutre, B Martino, S Saussele, HD Menssen, W Deng, N Krunic, V Bedoucha and G Saglio. Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the ENESTfreedom study. Leukemia (2017) 31, 1525â1531.
Dra. MarÃa Florencia Garaventa
Dto. BiologÃa Molecular
IBC Instituto de BioquÃmica ClÃnica